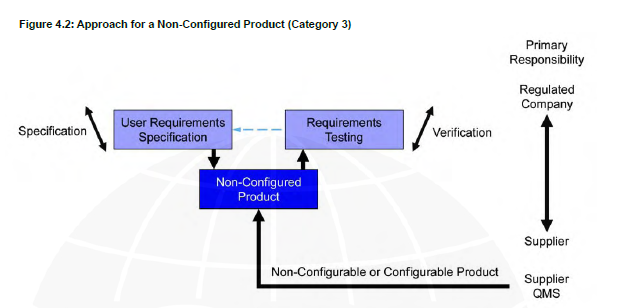
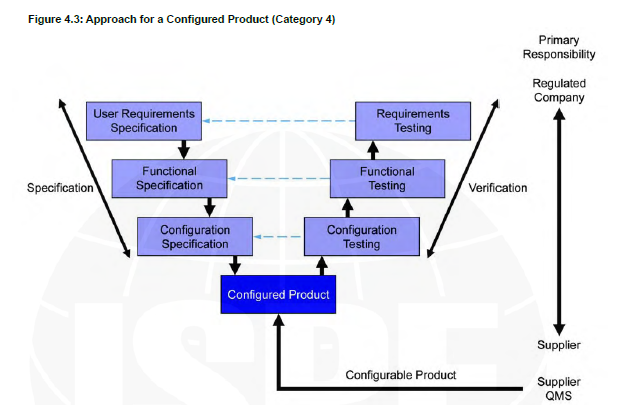
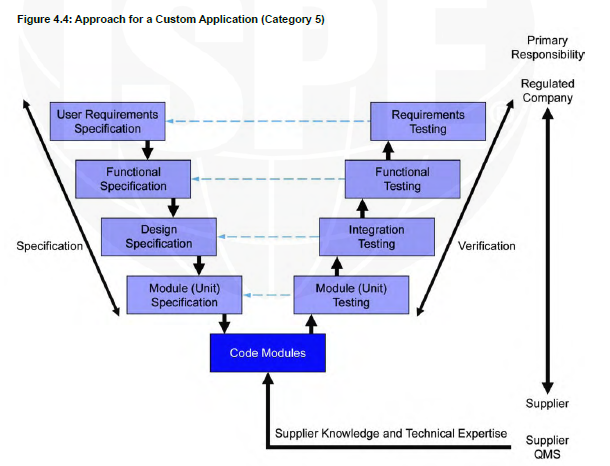
Computer system validation (CSV) is an essential process in the life science industry to ensure that computer systems used in the manufacturing, testing, and distribution of drugs are accurate, reliable, and secure. The guidelines provide a framework for the life cycle of computerized systems validation. The GAMP 5 is a set of guidelines established by the International Society for Pharmaceutical Engineering (ISPE) which is used by pharmaceutical and medical device manufacturers as well as suppliers to ensure compliance with regulatory requirements.
The following are some of the challenges of computer system validation:
▪ It can be a complex and time-consuming process.
▪ It requires a high level of expertise and knowledge.
▪ It can be expensive.
▪ It can be difficult to maintain compliance with regulatory requirements.
Despite the challenges, computer system validation is an essential process for ensuring the quality and safety of products and services.
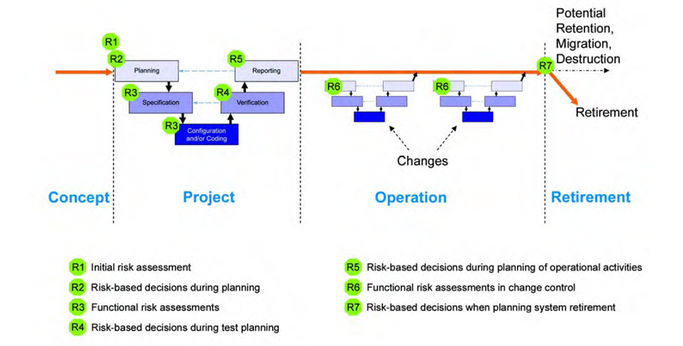
The life cycle of computerized systems validation, consists of four phases which are:
Concept:
▪ Define the system: The system should be defined in detail, including its purpose, scope, users.
▪ Identify risks: The system’s risks should be identified, including if its related to GxP and the questions we had in the priviest post.
Project:
🔸Planning
should cover all required activities, responsibilities, procedures, and timelines, life cycle activities should be scaled according to:
- System impact on patient safety, product quality, and data integrity (risk assessment)
- System complexity and novelty (architecture and nature of system components, including maturity and level of configuration or customization)
- Outcome of supplier assessment (supplier capability)
🔸Specification, Configuration and Risk assessment
Initial user requirements are often gathered during the concept phase and refined during the projected phase.
The role of specification is to enable systems to be developed, verified, and maintained. The number and level of detail of the specifications will vary depending upon the type of system and its intended use.
For example, software design specifications are not expected from the regulated company for non-custom products.
Specifications should be adequate to support subsequent activities, including risk assessment, further specification and development of the system, verification as appropriate, and system maintenance and update. The requirements for configuration and coding activities depend on the type of system (see Section 4.2.6 for examples).
Any required configuration should be performed in accordance with a controlled and repeatable process. Any required software coding should be performed in accordance with defined standards. The need for code reviews within the organization producing the software should be considered.
Configuration management and version control are intrinsic and vital aspects of controlled configuration and coding
🔸Verification
Verification confirms that specifications have been met. This may involve multiple stages of reviews and testing depending on the type of system, the development method applied, and its use.
Verification activities occur throughout the project stages.
Testing computerized systems is a fundamental verification activity. Testing is concemed with identifying defects so that they can be corrected, as well as demonstrating that the system meets requirements.
The use of effective and appropriate testing tools is encouraged. Such tools should have documented assessments for their adequacy (refer to EU Annex 11 Clause 4.7 [32]) and be controlled in use; however, validation is not required.
Testing is often performed at several levels depending on the risk, complexity, and novelty.
Tests may be defined in one or more test specifications to cover hardware, software, configuration, and acceptance.
🔸Report and Release
After the verification part of the project, we have the reports that approves that all verifications were completed, and that the system is approved for use.
The system should be accepted for use in the operating environment and released into that environment in accordance with a controlled and documented process. Acceptance and release of the system for use in GXP regulated activities should follow a defined process and involve oversight and input of the process owner, system owner, and the appropriate quality function as necessary and applicable.
A computerized system validation report should be produced summarizing the activities performed, any deviations from the plan, any outstanding and corrective actions, and provide a statement of fitness for intended use of the system.
The incorporation of lessons learned/after action review stage gates in the project should be considered (see /SPE
A well-managed system handover from the project team to the process owner, system owner, and operational users is a prerequisite for effectively maintaining compliance of the system during operation.
Traceability is recommended to add to the report to ensure that:
▪ Requirements are traceable back to business process needs
▪ Requirements are addressed and traceable to the appropriate functional and design elements in the specifications
▪ Requirements can be traced to the appropriate verification
As well as demonstrating coverage of design and verification, traceability can greatly assist the assessment and management of change.
Traceability should be focused on aspects critical to patient safety product quality, and data integrity.
Operation:
🔸Handover
Handover is the process for transfer of responsibility of a computerized system from a project team or a service group to the operations team or a new service group.
This is an important process; achieving compliance and fitness for intended use on its own may not be enough to guarantee a successful transfer into the operational phase.
The handover process will typically involve the project team), process owner, system owner, and Quality. The support group should be involved at the earliest opportunity to ensure effective knowledge transfer and establishment of operational procedures. A period of elevated support and maintenance, often referred to as Hypercare Services, may be arranged to facilitate the transfer.
Implied context-specific knowledge acquired through personal experience or internalization, as well as explicit knowledge captured and codified in documentation, tools, and systems should be considered.
🔸Service Management and Performance Monitoring
Service management and performance monitoring are shown related to records management due to records generated to demonstrate proper operation and performance of a system. In addition, there is potential interaction with incident and problem management and CAPA and change management when the results of the service or monitoring indicate there are issues that need addressing.
- Establishing and Managing Support Services
The support required for each system, and how it will be provided, should be established. Support may be provided by external service providers or internal resources. This process should ensure the following:
▪ Service agreements
▪ Maintenance plans
▪ SOPs
▪ Support systems
2. Performance Monitoring
Performance monitoring detects issues that could impact the availability and performance of the system in order to facilitate mitigation before problems occur. Detected issues are managed through the incident management and problem management processes. Monitoring tools and automation are increasingly used to detect potential issues, report issues, and escalate to support organizations for timely intervention and rectification, and are encouraged.
The need for performance monitoring should be considered, and required activities scheduled and documented. This may change during the operational life of a system.
🔸Incident and Problem Management and CAPA
- Incident Management and Problem Management
The incident management process aims to categorize incidents to direct them to the most appropriate resource or complementary process to achieve a timely resolution; whereas problem management involves analyzing root causes and preventing incidents from happening in the future.
There should be a procedure defining how problems related to software, hardware, and procedures should be captured, reviewed, prioritized, progressed, escalated, and closed.
This includes the need for processes to monitor progress and provide feedback.
Incident and problem management processes may be supported by service management tools that support the incident and problem management process, associated action planning, and traceability of actions taken to address the incident or problem.
🔸Corrective and Preventive Action
CAPA is a process for investigating, understanding, and correcting discrepancies based on root-cause analysis, while attempting to prevent their recurrence.
In the operational environment the CAPA process should feed into the overall CAPA system used for GxP operations.
When incidents occur, or when opportunities to reduce process/system failures are identified by other means, CAPA should be identified and processes established to ensure that these are implemented effectively.
🔸Change Management
- Change Management
Change management is a critical activity that is fundamental to maintaining the proper functioning and controlled status of systems and processes. All changes that are proposed during the operational phase of a computerized system, whether related to software (including middleware), hardware, infrastructure, or use of the system, should be subject to a formal change-control process. This process should ensure that proposed changes are appropriately reviewed to assess impact and risk of implementing the change.
Regression analysis and regression testing may be required. The process should ensure that changes are suitably evaluated, authorized, documented, tested, and approved before implementation, and subsequently closed.
The process should allow the rigor of the approach, including the extent of documentation and verification, to be scaled based on the nature, risk, and complexity of the change, by application of critical thinking. Some activities such as replacements and routine system administration tasks should be covered by appropriate repair or system administration processes.
Change management should provide a mechanism for prompt implementation of continual process and system improvements based on periodic review and evaluation, operational and performance data, and root-cause analysis of failures.
🔸Configuration Management
Configuration management includes those activities necessary to precisely define a computerized system at any point during its life cycle, from the initial steps of development through to retirement.
A configuration item is a component of the system that does not change as a result of the normal operation of the system. Configuration items should only be modified by application of a change management process. Formal procedures should be established to identify, define, and baseline configuration items, and to control and record modifications and releases of configuration items, including updates and patches.
🔸Repair Activity
The repair or replacement of defective computerized system components, which are often but not exclusively hardware or infrastructure related, should be managed in accordance with a defined process. Such activities should be authorized and implemented within the wider context of the change management process. Many repair activities are emergencies and require rapid resolution, so the incident and change management processes should be designed to allow such activities to occur without delay or increased risk to the operational integrity of the computerized system.
🔸Periodic Review
Periodic reviews are used throughout the operational life of systems to verify that they remain compliant with regulatory requirements, fit for intended use, and meet company policies and procedures, including those related to data integrity. The reviews should confirm that, for components of a system, the required support and maintenance processes and expected regulatory controls (plans, procedures, and records) are established.
Periodic reviews should be:
- Scheduled at an interval appropriate to the impact and operational history of the system. Risk and other assessments should be used to determine which systems are in scope and the frequency of periodic review.
- Performed in accordance with a predefined process
- Documented with corrective actions tracked to satisfactory completion
🔸Continuity Management-
- Backup and Restore
Processes and procedures should be established to ensure that backup copies of software, records, and data are made, maintained, and retained for a defined period within safe and secure areas
Restore procedures should be established, tested, and the results of that testing documented.
🔸Business Continuity Planning
Business Continuity Planning (BCP) is a series of related activities and processes concerned with ensuring that an organization is fully prepared to respond effectively in the event of failures and disruptions, covering local and global infrastructure, data, and the application
Critical business processes and systems supporting these processes should be identified, and the risks to each assessed. Plans should be established and exercised to ensure the timely and effective resumption of these critical business processes and systems
A business continuity plan defines how the business may continue to function and handle data following failure. It also defines the steps required to restore business processes following a disruption and, where appropriate, how data generated during the disruption should be managed. Plans should be prioritized based on patient and business risk, in case of disruption to multiple systems.
The BCP also identifies the triggers for invoking the business continuity plan, roles and responsibilities, and required communication.
🔸Disaster Recovery Planning
As a subset of BC, plans should be specified, approved, and rehearsed for the recovery of specific systems in the event of a disaster. These plans should detail the precautions taken to minimize the effects of a disaster, allowing the organization to either maintain or quickly resume critical functions. There should be a focus on disaster prevention, e.g., the provision of redundancy for critical systems. Disaster Recovery (DR) plans often require a shared responsibility model between internal organizations of the regulated company and external service providers.
🔸Security and System Administration-
- Security Management
Computerized systems and data should be adequately protected against willful or accidental loss, damage, or unauthorized change. Procedures for managing secure access, including adding and removing privileges for authorized users, virus management, password management, and physical security measures should be established before the system is approved for use
Role-based security should be implemented, if possible, to ensure that sensitive data and functions are not compromised. Security management procedures should apply to all users, including administrators, superusers, users, and support staff (including supplier support staff).
Security provisions should ensure that data is protected against unauthorized intrusions including cyber security attacks. Intrusion prevention and detection processes, supported by relevant IT tools and automation, should be in place.
🔸System Administration
Once a system is in operation the users of the system will require support. The system administration process provides administrative support for systems, including performance of standard administration tasks. The extent of this process varies greatly depending on the nature of the system.
🔸Record Management
- Retention
Policies for the retention of regulated records should be established, based on a clear understanding of regulatory requirements and existing corporate policies, procedures, standards, and guidelines.
🔸Record Management- Archiving and Retrieval
Archiving is the process of taking records and data off-line by moving them to a different location or system, often protecting them against further changes. Procedures and processes for archiving and effective and accurate retrieval of records should be established based on a clear understanding of regulatory requirements including retention periods.
Retirement:
▪ Retire the system: The system should be retired from use when it is no longer needed.
▪ Archive the data: The system’s data should be archived.
This part covers system withdrawal, system decommissioning, system disposal, and migration of required data.
🔸Withdrawal
Withdrawal is the removal of the system from active operation, i.e., users are deactivated, interfaces disabled. No data should be added to the system from this point forward. Special access should be retained for data reporting, results analysis, and support.
🔸Decommissioning
Decommissioning is a controlled process by which an application or system is removed from use in an organization once it has been retired and/or has become obsolete.
🔸Disposal
Data, documentation, software, or hardware may be permanently destroyed. Each may reach this stage at a different time. Data and documentation may not be disposed of until they have reached the end of the record-retention period, as specified in the company’s record-retention policy, following an authorized and documented process.
Due to the volume of data and records involved, retirement can be a major task for IT systems in particular.
Consideration should be given to:
- Establishing procedures covering system retirement, including withdrawal, decommissioning, and disposal as appropriate
- Documentary evidence to be retained of actions taken during retirement of the system
- GxP records to be maintained, their required retention periods, and which records can be destroyed
- The need to migrate records to a new system or archive, and the method of verifying and documenting this process
- Ability to retrieve these migrated records on the new system
🔸Data Migration
Data migration may be required when an existing system is replaced by a new system, when an operational system experiences a significant change, or when the scope of use of a system is changed. The migration process should be accurate, complete, and verified.
In conclusion, the GAMP5 guidelines provide a framework for the life cycle of computerized systems validation which is essential in ensuring that computer systems used in the Life Science industry are accurate, reliable, and secure. The five phases of the life cycle, namely planning, risk assessment, specification, testing, and maintenance, provide a structured approach to the validation process, which helps to ensure compliance with regulatory requirements.

Rafael Port
Validation & Engineering Project Manager