Targeted cancer therapy continues to evolve, and many new treatment combinations are being approved in addition to developmental advancements in novel checkpoint inhibitors. This article will address the lately approved antibody therapeutics (US and EU), those in regulatory review, the expanded approval of existing immunotherapies, and their associated companion diagnostics for cancer treatment. This article will address the following issues:
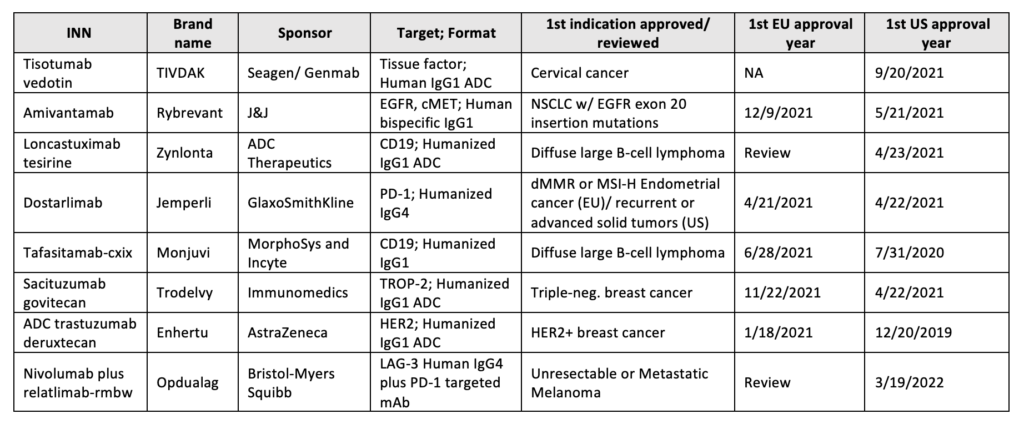
Between 2021 to 2022, the FDA approved six new antibody-based immunotherapies and EMA five whereas four of the five accepted were previously cleared by the FDA, Table 12,3.
Table 1: Antibody therapeutics approved in the EU or US
Starring 2021’s approval is Dostarlimab (Jemperli), anti-PD-1 Humanized IgG4, and Amivantamab (Rybrevant), a bispecific antibody targeting cancer epitopes, EGFR and MET. Both were approved in the EU (M.A. No. EU/1/21/1538/001) and US (BLA:761174/761210) to treat tumors with a high mutational burden and NSCLC with EGFR exon 20 mutations, respectively.
Companion Diagnostics and Future Artificial Intelligence (AI)-Based Approaches
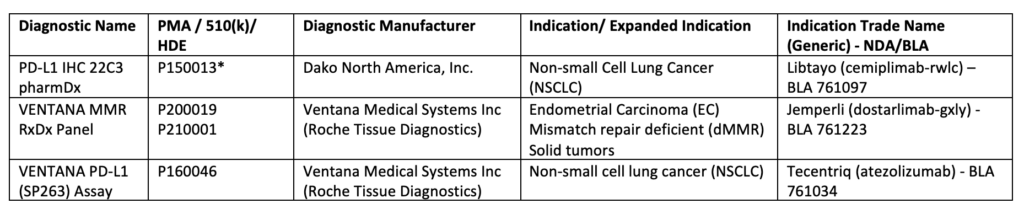
The use of immune signature to predict immunotherapeutic response led to an increase in the development and approval of immunohistochemical (IHC) diagnostic assays, Table 24. Such devices allow informative decision-making to assess tumor-favoring immune responsiveness5,6. For example, the VENTANA MMR RxDx panel was approved concomitantly with Dostarlimab (mentioned above) to distinguish the patients with dMMR who will benefit from this treatment7. Additional FDA-approved companion diagnostic tests are listed below, Table 2.
Table 2:List of Approved Companion Diagnostic Devices
Important past year’s (2021) step in the immunotherapeutic approach is the expanded indication approvals for several already in-use drugs as adjuvant/neo-adjuvants therapies for the core axis PD-1/PD-L1. PD-1/PD-L1 blockade was proven to promote DFS (Disease-Free Survival) and EFS (Event-Free Survival). Atezolizumab (Tecentriq) had been cleared by the FDA as an adjuvant treatment with the companion diagnostic test VENTANA PD-L1 (SP263), mentioned above, for NSCLC (non-small-cell lung cancer) patients expressing PD-L113. Pembrolizumab (Kytruda) was approved both by EMA and FDA for the adjuvant treatment of RCC (Renal Cell Carcinoma) and neoadjuvant+adjuvant treatment for TNBC (Triple Negative Breast Cancer), respectively 14,15 . Nivolumab(Opdivo) has also been approved by the E.C. (European Commission) and the FDA for the adjuvant treatment of esophageal or GEJ (gastroesophageal junction) cancer16,17. The latter had also approved Nivolumab for adjuvant therapy of urothelial carcinoma18.
Latest Advancements in the Immunotherapy Field and 2022 Expected Approvals
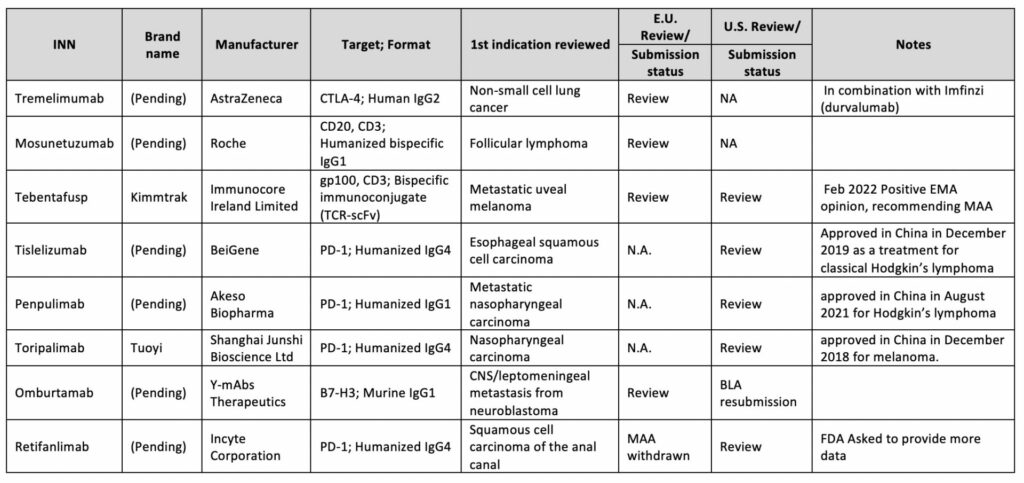
From a regulatory point of view, the unexpected immune response of antibody-based biopharmaceuticals is one of the obstacles when developing biological products. Thus it is advised to navigate the strategic and regulatory decisions correctly to properly assess the safety and efficacy of such biotherapeutic 22. These assessments are usually conducted during pivotal clinical trials; as per ICH S6(R1), nonclinical immunogenicity data are not considered to be predictive of clinical safety 23. Even a greater challenge arises when designing antibody-based immunotherapy with expansion to dual targets i,e. Bispecific Antibodies (bsAbs). Such a strategy may increase immunogenicity issues which may also be related to novel complex structures. Consequently, for benefit-risk assessment, the FDA may request a comparison of the bsAb to an approved monospecific product directed against the same antigenic target(s)24.
In any circumstance, every applied BLA needs to show a clinical advantage. For example, FDA denied the approval of Retifanlimab (PD1-targeted mAb)25and Oportuzumab monatoxalso (EpCAM-targeted immunotoxin, ADC), questioning their clinical benefit. In addition, concerns were raised about the Oportuzumab monatoxalso company’s CMC (Chemistry, Manufacturing, and Controls) processes26.
Accelerated approvals are granted to many I.O. biologics; however, some are not reviewed for years. In 2021, FDA conducted an industry-wide evaluation of such drug approvals, which failed to meet post-marketing requirements 27,28. Following FDA’s call into question, several immunotherapy indications were withdrawn and removed from the US market28,29,30.
1. Cox EM, Edmund A V, Kratz E, Lockwood SH, Shankar A. Regulatory Affairs 101 : Introduction to Expedited Regulatory Pathways. 2020;2012:451-461. doi:10.1111/cts.12745
2. Www.antibodysociety.org.17Dec2021. Antibody therapeutics approved or in regulatory review in the EU or US. Antib Soc. Published online 2021. https://www.antibodysociety.org/resources/approved-antibodies/
3. Mullard A. 2021 FDA approvals. Nat Rev Drug Discov. 2022;(January). doi:10.1038/d41573-022-00001-9
4. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). doi:https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools
5. Emily A. Prince, PharmD1 ; Jenine K. Sanzari, PhD1 ; Dimple Pandya, MD1 ; David Huron P; and RE. Analytical Concordance of PD-L1 Assays Utilizing Antibodies From FDA-Approved Diagnostics in Advanced Cancers : A Systematic Literature Review abstract. Published online 2022. doi:10.1200/PO.20.00412
6. Christian U. Blank, John B. Haanen AR and TN. The cancer immunogram. Science (80- ). 2016;352(6286):658-660. doi:10.1126/science.aaf2834
7. VENTANA MMR RxDx Panel – P200019. Published 2021. https://www.fda.gov/medical-devices/recently-approved-devices/ventana-mmr-rxdx-panel-p200019
8. Baxi V, Edwards R, Montalto M, Saha S. Digital pathology and artificial intelligence in translational medicine and clinical practice. Mod Pathol. 2022;35(1):23-32. doi:10.1038/s41379-021-00919-2
9. Xu Z, Wang X, Zeng S, Ren X, Yan Y, Gong Z. Applying artificial intelligence for cancer immunotherapy. Acta Pharm Sin B. 2021;11(11):3393-3405. doi:10.1016/j.apsb.2021.02.007
10. Artificial Intelligence and Machine Learning in Software as a Medical Device_Action Plan. https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-software-medical-device
11. Good Machine Learning Practice for Medical Device Development: Guiding Principles. https://www.fda.gov/medical-devices/software-medical-device-samd/good-machine-learning-practice-medical-device-development-guiding-principles
12. Roche announces the release of its newest artificial intelligence (AI) based digital pathology algorithms to aid pathologists in evaluation of breast cancer markers, Ki-67, ER ,and PR. https://diagnostics.roche.com/global/en/news-listing/2021/roche-announces-release-of-its-newest-ai-based-digital-pathology-algorithms-to-aid-pathologists-in-evaluation-breast-cancer-markers-ki67-er-pr.html
13. FDA F and DA. FDA approves atezolizumab as an adjuvant treatment for non-small cell lung cancer. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-adjuvant-treatment-non-small-cell-lung-cancer
14. FDA F and DA. FDA approves pembrolizumab for high-risk early-stage triple-negative breast cancer. Published 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-high-risk-early-stage-triple-negative-breast-cancer
15. EMA. Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion for the extension of Kytruda as monotherapy adjuvant treatment_RCC. Published 2021. https://www.ema.europa.eu/en/medicines/human/summaries-opinion/keytruda-6
16. Bristol Myers Squibb. Bristol Myers Squibb Receives European Commission Approval for Opdivo (nivolumab) as Adjuvant Treatment for Esophageal or Gastroesophageal Junction Cancer Patients with Residual Pathologic Disease Following Chemoradiotherapy. Published 2021. https://news.bms.com/news/corporate-financial/2021/Bristol-Myers-Squibb-Receives-European-Commission-Approval-for-Opdivo-nivolumab-as-Adjuvant-Treatment-for-Esophageal-or-Gastroesophageal-Junction-Cancer-Patients-with-Residual-Pathologic-Disease-Following
17. Bristol Myers Squibb. U.S. Food and Drug Administration Accepts for Priority Review Application for Opdivo® (nivolumab) as Adjuvant Therapy for Patients with Resected Esophageal or Gastroesophageal Junction Cancer. Published 2021. https://news.bms.com/news/details/2021/U.S.-Food-and-Drug-Administration-Accepts-for-Priority-Review-Application-for-Opdivo-nivolumab-as-Adjuvant-Therapy-for-Patients-with-Resected-Esophageal-or-Gastroesophageal-Junction-Cancer/default.aspx
18. FDA F and DA. FDA approves nivolumab for adjuvant treatment of urothelial carcinoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-nivolumab-adjuvant-treatment-urothelial-carcinoma
19. Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13(1). doi:10.1080/19420862.2020.1860476
20. Si Y, Melkonian AL, Curry KC, et al. Monoclonal antibody-based cancer therapies. Chinese J Chem Eng. 2021;30:301-307. doi:10.1016/j.cjche.2020.11.009
21. Vaddepally RK, Kharel P, Pandey R, Garje R. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. :1-19.
22. Vandivort TC, Horton DB, Johnson SB. Regulatory and strategic considerations for addressing immunogenicity and related responses in biopharmaceutical development programs. J Clin Transl Sci. 2020;4(6):547-555. doi:10.1017/cts.2020.493
23. European Medicine Agency. EMA/CHMP/ICH/731268/1998 ICH guideline S6 (R1) on preclinical safety evaluation of biotechnology-derived pharmaceuticals. Comm Med Prod Hum use ICH. 2011;6(June):1-22. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-s6r1-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals-step-5_en.pdf
24. Gough J, Nettleton D. Bispecific Antibody Development Programs – Guidance for Industry_CEDER. Manag Doc Maz. 2019;(April):10.
25. Tucker N. FDA Denies Approval of Retifanlimab for Locally Advanced or Metastatic SCAC Subgroup. Published 2021. https://www.targetedonc.com/view/fda-denies-approval-of-retifanlimab-for-locally-advanced-or-metastatic-scac-subgroup
26. Biospace. FDA Slams Sesen Bio with CRL for Bladder Cancer Drug. Published 2021. https://www.biospace.com/article/fda-crushes-sesen-bio-with-crl-for-bladder-cancer-drug/
27. Astor L. FDA Cracks Down on Dangling Accelerated Approvals in 2021, Pathway Is Scrutinized. Published 2021. https://www.targetedonc.com/view/fda-cracks-down-on-dangling-accelerated-approvals-in-2021-pathway-is-scrutinized
28. FDA In Brief: FDA Oncologic Drugs Advisory Committee to Review Status of Six Indications Granted Accelerated Approval. Published online 2021. https://www.fda.gov/news-events/fda-brief/fda-brief-fda-oncologic-drugs-advisory-committee-review-status-six-indications-granted-accelerated
29. Merck withdraws Keytruda from SCLC indication amid FDA crackdown. Published 2021. https://www.clinicaltrialsarena.com/news/merck-withdraws-keytruda-for-lung-cancer-amid-fda-crackdown/
30. Nivolumab Indication in Small Cell Lung Cancer Withdrawn in U.S. Market. Published 2021. https://ascopost.com/issues/january-25-2021/nivolumab-indication-in-small-cell-lung-cancer-withdrawn-in-us-market/
This article was prepared by:
Vered Ben Hur. Ph.D
Pharma Regulation Projects Manager
For more information about our Pharmaceuticals industry visit:
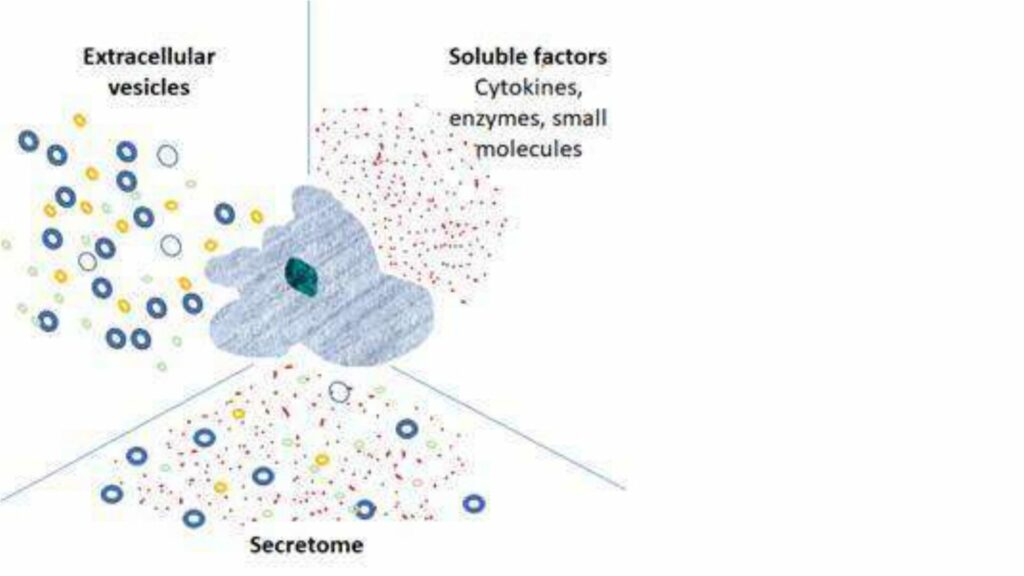
In the last decade, the cell therapy field has matured, with some notable successes, including CAR-T therapies and gene-modified cell therapies targeting specific rare diseases. There have also been some disappointments, in particular, the failure of several MSC therapies to reach efficacy endpoints in late-stage clinical trials. In parallel, however, the realization that very many beneficial effects of cell therapy can be attributed to the cells’ secretome, has gained traction. This is evident in the exponential increase in publications relating to the secretome and extracellular vesicles (EVs), as well as in the number of start-up companies engaged in the development of such cell-free cell-derived products.
Enter the new kids on the block: cell-derived, cell-free products.
These products are derived from cell culture’s secretions – the secretome – which includes: soluble proteins and peptides, cytokines, chemokines, lipids, carbohydrates, lipid bi-layer bound vesicles including micro-vesicles, extracellular-vesicles (EVs), small EVs (sometimes referred to as exosomes) and their contents, which in addition to the above, contain nucleic acids (DNA and miRNA). The composition of the secretome depends on the cell type and microenvironmental stimuli. This is a dynamic and rich soup!
In addition, to a cell’s natural secretions, companies are engineering and/or activating cells to specifically influence the secretome by, for example, enriching for secretion of a particular factor, or directing expression of a particular factor to the surface of an EV.
What are the advantages of EV’s and Secretome – cell-free cell-derived products?
Cell-free cell-derived products may offer a number of advantages over cell therapies:
In terms of manufacturing – such a product is likely to be more stable, possibly amenable to lyophilization ,and may not require cryopreservation. This represents a huge benefit in terms of process timing, testing ,and transport logistics compared to cell therapies.
In terms of safety – a cell-free product will have no persisting cells and therefore, no potential for transformation and risk of tumor formation. Similar to traditional biological products, an EV or secretome-based product is expected to have a relatively short-term effect with half-life and clearance by predictable pathways.
In terms of efficacy – a cell-free product containing multiple active components enables a broad-spectrum approach to multiple targets, amenable for a range of indications. Repeat dosing is more likely to be required, but dosing should be easier to tailor to the patient’s requirements, based on traditional PK understandings.
What are the main hurdles to be overcome when developing such a product?
One of a kind
All cell-free cell-derived products we’ve encountered at Gsap have been very different from each other in terms of the manufacturing process, product description ,and indication. As with cell therapy, each product is unique with the manufacturing process and controls developed to suit each individual product, and careful risk assessment, is a must, to accompany each step in the development.
Cell banking
Of particular importance is the use of a well-characterized, fully tested cell bank, suitable for GMP manufacture. The cell bank is subject to the same rigorous testing requirements as required for cell therapy. The use of well-characterized commercially available cell banks, manufactured according to GMP, with a well-documented history and testing certification can be of great benefit to enable efficient translation of cell-free cell-derived products into the clinic.
Manufacturing process
Similar to cell therapy, the concept that the product is the process certainly applies here. The scientific literature is replete with examples illustrating the impact of the environment, culturing conditions, cell source, cell type, donor age, media, reagents, scale-, and vessel type, amongst other things, on cell secretome content. Early implementation of appropriate process controls is, therefore, essential to develop a process for a product sensitive to so many variables. EV or secretome-based products may be less potent than cell therapy products unless the manufacturing process can successfully concentrate the main active components whilst eliminating impurities.
No benchmarks
To date, there are no approved EV or secretome-based products approved for marketing in any territory. In contrast, the main risks from cell therapy have been established, over almost two decades of clinical use. In the absence of any benchmark products and potentially more risks, it is critical to generate an understanding for the development product’s mechanism of action (MOA) in order to successfully mitigate potential risks.
Regulatory considerations
Combined regulations are expected to apply to cell-free cell-derived products such as EVs and secrotome products, both in the US and in Europe. The exact product classification may vary, based on the product composition and attributes.
Traditional biological product regulations are expected to apply with regard to nonclinical safety testing, in other words, unlike cell therapy requirements, PK and safe pharmacology evaluation may be required and GLP safety studies may be required in two species. Safety and toxicology programs should be agreed with the regulator in advance. With cell-derived products, the potential for confounding immune response should be carefully considered.
Conclusions
These are exciting times, as biotech companies explore the use of cell secretomes for therapeutic benefit. Such cell-free cell-derived products present a whole new list of challenges for the developer and regulator alike. The new products in development are complex mixtures that require sophisticated analytical methods for their characterization – proteomics, nanoparticle tracking analysis, next-generation sequencing ,and mass spectrometry are just a few of the methods that we can expect to see more of. As with cell therapy, we trust that the individual regulatory bodies will harmonize their requirements so that development is not needlessly complicated further.
This article was prepared by both:
Diana Gershtein MSc., MBA
Advanced Therapies Section Manager
Tami Horovitz, PhD
Gsap Regulatory Submissions – Content Expert
For more information about our Pharmaceuticals industry visit: