As many of you know, the European Union is going through a significant change in the regulation of medical devices. First was the MDD to MDR, and now is the time for the IVDD to transition into the IVDR. This transition has a significant effect on many of the IVD products that will have to go through the new certification process.
This article will review some of these changes:
2- How are IVDs different from medical devices?
3- The European Union (EU) is beginning a new era
4- Timelines
5- Clinical-Evidence/-Performance-Evaluation
What are IVD’S?
‘In vitro, diagnostic medical device’ means any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally to provide information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.
Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices; [Article 2 2017/746]
In simple words, IVDs are medical devices and accessories used to perform tests on samples, such as blood, urine, tissue is taken away from the human body to help detect infection, diagnose a medical condition, prevent disease, or monitor drug therapies.
Some examples of IVDs include systems to test the level of coagulation, urine test strips, home use pregnancy tests, HIV or Hepatitis tests, blood glucose monitoring, etc. IVD device may be used by laypeople (home-use blood glucose level) or by a health professional (detection of N. meningitidis in CSF or blood)
How are IVDs different from medical devices?
An IVD is a subset of a medical device. The legislator created this subcategory and made different requirements for the IVD due to the different risk sets associated with them: no direct contact with the patient, the value of the medical data they deliver, and that they don’t provide treatment.
IVDs fulfill their role based on information they provide, without direct contact with the patient or direct action on the patient. This is of fundamental importance and significantly impacts product validation and testing.
The quality of the information delivered by an IVD is assessed by measuring the analytical precision of the test or assay, and the clinical evidence of the information provided. It is evident how important it is for the IVD product to be very precise, for example, when testing blood for blood-typing (identifying blood type) or screening for HIV, Covid 19, etc. We also understand the clinical relevance of these assays – this is why those IVDs with the highest risk potential on the population must have exact performance criteria and results.
The European Union (EU) is beginning a new era
In Europe, the field of IVD has been controlled by the IVD Directive (IVDD) 98/79/EC that was issued in 1998, and compliance became mandatory in Dec 2003. This era is coming to an end as we are shifting to the new generation of the IVD Regulation (IVDR) 2017/746 published on April 5th, 2017, which is currently beginning to come into effect.
The old directive was relatively descriptive, while the new regulation is very prescriptive. To illustrate, the IVDD is 37pages, whereas the IVDR is 204pages. The IVDR was written in the same spirit as the MDR regulation and they have a lot of similarities.
Some of the main points in the IVDR:
●Risk-based classification
●Classification categories were changed.
●Product whole lifecycle
●New subcategories of IVDs (Devices for near-patient testing, Companion diagnostic, Software devices)
●Greater scrutiny of Notified Bodies
●No “grandfathering” provisions. All currently approved IVD devices must be recertified under the new requirements and demonstrate they are state of the art.
●More rigorous clinical evidence. Manufacturers need to conduct clinical performance studies and provide evidence of safety and performance according to a device’s assigned risk class.
●Requirements for post-market surveillance were significantly increased, and the general timeline for reporting was reduced.
●General Safety and Performance Requirements (GSPR) replaced the “essential requirements”
●Identification of ‘person responsible for regulatory compliance.’ (PRRC)
●Implementation of unique device identification (UDI) for better traceability and recall (See UDI newsletter in Gsap web-site)
●More responsibility for economic operators, etc.
Timelines
The IVDR 2017/746 entered into force in May 2017, with a final Date of Application of May 2022 for new devices, with a progressive rollout for devices with a valid IVDD certificate.
However, all IVD products on the market after 26 May 2022 must comply with the IVDR for post-market surveillance, market surveillance, vigilance, registration of economic operators, and devices. [2017/746 article 113]
When talking about the progressive rollout, one must understand some key definitions:
Placing on the Market; [2017/746 (21)] it means to supply the product to a distributor or the end-user, not the importer.
Putting into Service [2017/746 (22)] means the first time the product is used commercially and not in a study.
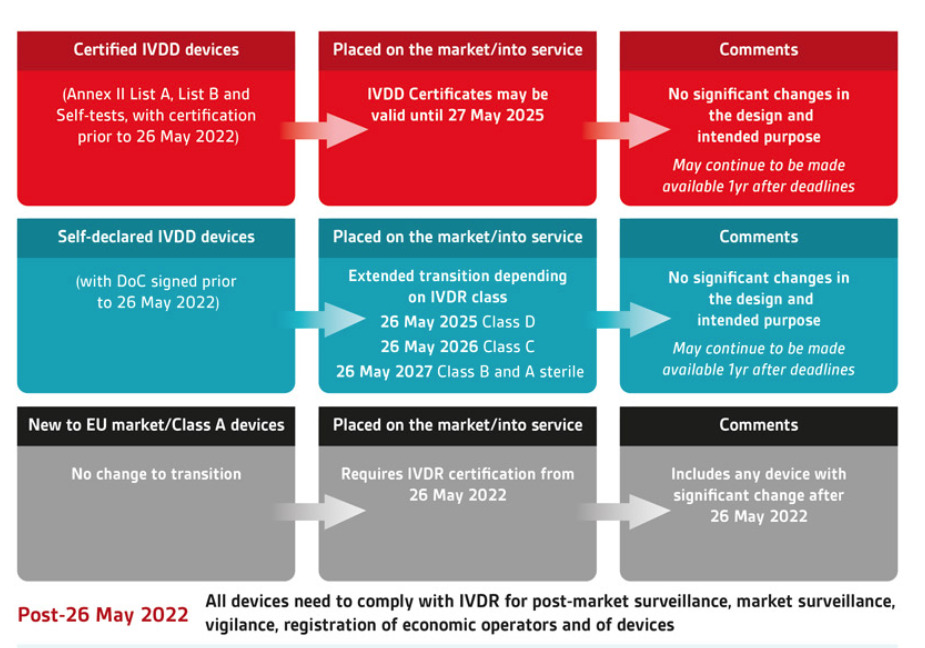
The picture above illustrates how the old and new classification, together with the definition mentioned above (placing on the market, putting into service), will affect the deadline to:
● When new products cannot be introduced into the market
● When inventory cannot be sold from the manufacturer/ importer to the distributor
● When inventory cannot be used
as you can see, the best-case products that are self-certified and are class A or B under the new classification of the IVDR, can have their products used up to May 2028.
Classification of IVD:
IVDD classification is “list-based” and divided into four categories: (from the lowest to the highest risk): General, self-test, list B (annex II), list A (annex II). IVDR is Risk-based and divided into four categories (from the lowest to the highest risk) Class A, A sterile, B, C, D
IVDR classification is based on seven rules; one should go over each rule and see if it applies to its device; the NB wants to know the process of classification, and so you need to address each rule that applies, then you indicate which is the highest class/ highest risk and that’s the rules that apply.
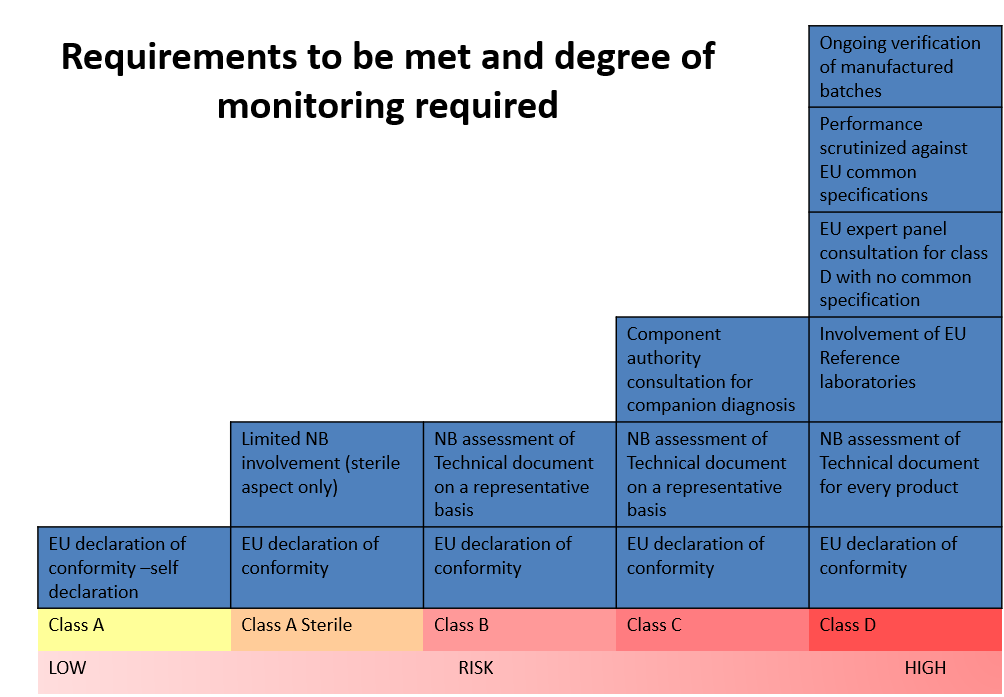
As demonstrated in the picture above, there is a distinct correlation between the class of the device (risk) and the degree of regulatory requirements and the NB monitoring.
This change in classification has a significant impact on most IVD devices. Under the IVDD, there are about 40,000 devices. Of them, only 8% require NB certification; however, it is estimated that under the IVDR classification, 78% of the devices would need NB certification. Furthermore, it is estimated that about 22% of devices will not seek accreditation under IVDR due to the increased cost. [2] Products sold under FDA approval may find that they have a leg up in gathering the necessary information.
| Distribution of IVD devices based on the IVDD classification |
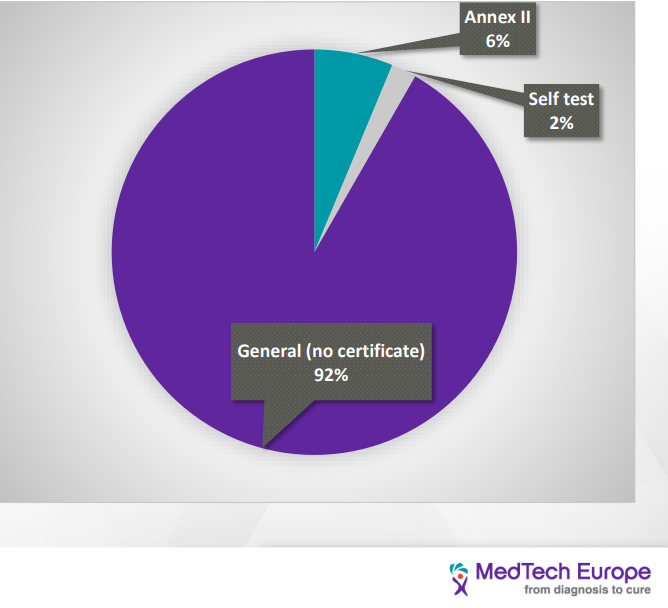
| Distribution of IVD devices based on the IVDR classification |
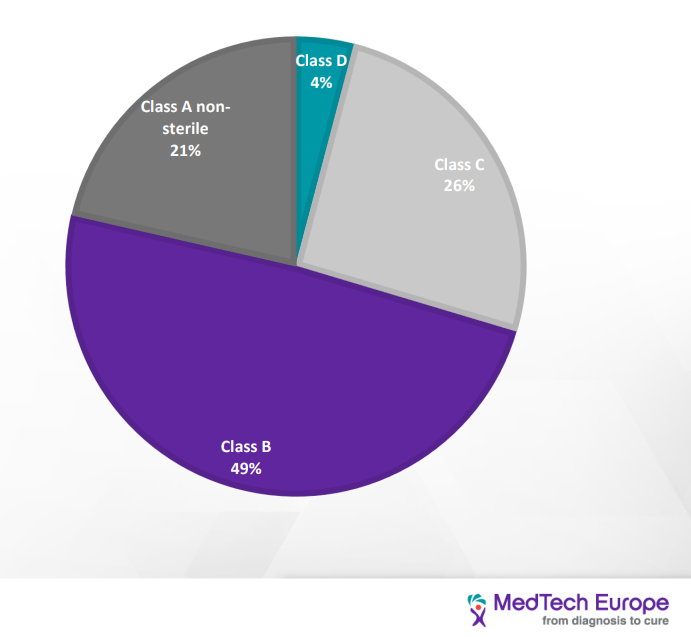
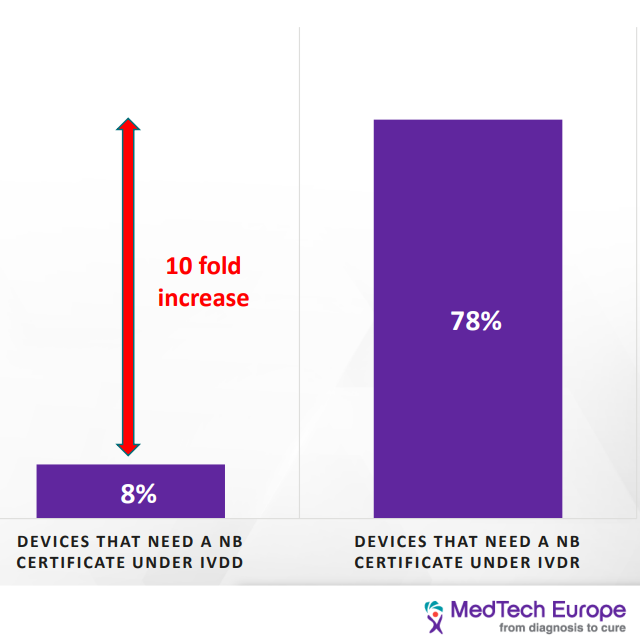
| As we can see, the load on the NB is going to increase tenfold due to the increasing number of devices that would seek certification, and at the same time, the amount of NB qualified to provide certificates for the IVDR has decreased from twenty-one to six. This would have a significant impact on the NB availability and timeline to process new devices. Current turnaround time is 9-11 month and go to be as long as 18 months. [3] |
The change in classification method will drastically change the class of many devices, as demonstrated in the illustration above and by so, also the number of regulatory requirements to comply with. For example, COVID 19 self-test, is classified as list B under the IVDD. In contrast, under the IVDR, it will be class D. Another example is the COVID 19 PCR test that under the IVDD is self-certified were as under the IVDR, it is class D (highest risk category).
The best practice is first to figure out the new classification of one’s device, which will allow you to know the regulatory requirements, perform a gap assessment and create a plan to close the gaps.
Clinical Evidence/ Performance Evaluation
Since the IVDs don’t have direct contact with the patient and don’t impact the patient health directly, there is no clinical evaluation but performance evaluation.
BSI Data revealed that 40% of the first round of questions were about clinical performance. The next topic was in regards to analytical performance. [4]
The performance evaluation for IVD is composed of three pillars:
- Scientific validity-
means the association of an analyte with a clinical condition or a physiological state; 2017/746 (38) - Analytical performance-
means the ability of a device to correctly detect or measure a particular analyte; 2017/746 (40). This may be demonstrated with the following test: sensitivity, specificity, precision, accuracy, the limit of detection, cut off, etc. - Clinical performance-
the ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user. 2017/746 (41)
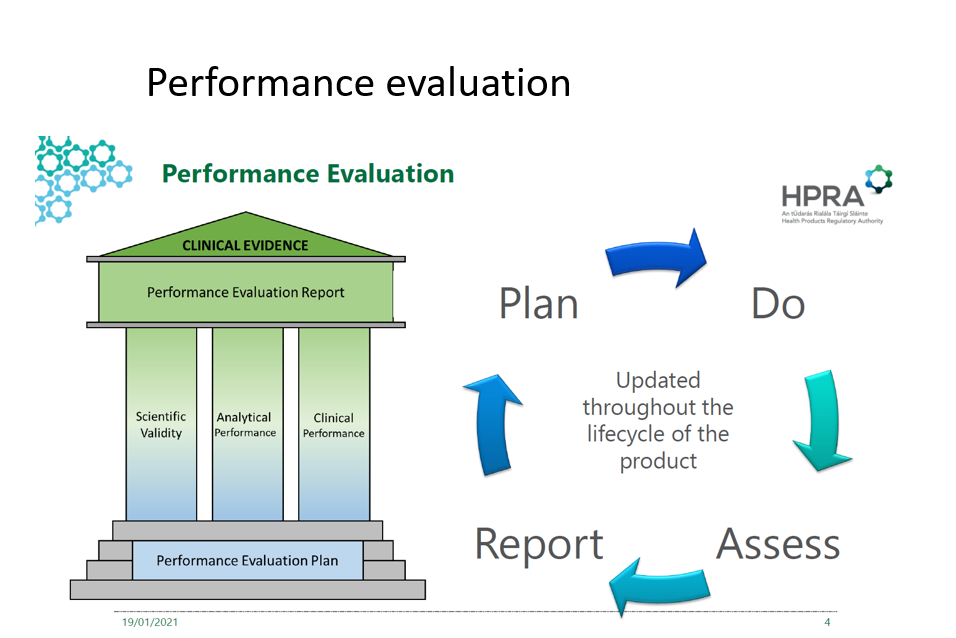
The picture above demonstrates the continuous cycle of performance evaluation and its pillars.
State of the Art:
Means what is currently regarded as good practice, not the most advanced technology [ISO/IEG Guide 63:2019, 3.18]
Performance evaluation aims to scientifically demonstrate that the device is safe, state of the art and that the intended clinical benefit is achieved. This can and should be done by relevant study data, literature, and other sources of technical information and the analysis and conclusions of the performance evaluation process.
Seen as the new regulation has put a big emphasis on the whole lifecycle of the device, the manufacturer is required to demonstrate the info for the certification and then continuously and actively gather this information throughout the whole lifecycle of the device, together with the post-market surveillance (PMS).
A few tips that we learned along the way:
● Start sooner rather than later, it’s a long unfamiliar process, and the queue is getting longer by the minute.
● The NB wants to see the IVDR jargon used.
● Under the IVDR, you only get three rounds of questions from the NB, and then you have to restart (Unlike in the IVDD that you could go back and forth with NB)
● Whenever you don’t perfume a requirement since it’s not applicable, you must provide a justification
● Provide an extensive glossary and acronyms (the reviewer may be a health professional but not necessarily from the industry).
● Make sure your crucial narrative/stories are clear, that each document has a beginning, middle, and end, and summaries of the conclusions.
● Be consistent! Review for consistency in the document and across all the documents for the same names, terms, definitions, etc.
This article was prepared by:

Dr. Tami Siniaver
RA & QA Project manager, Medical device